“A novel PAX3 mutation in a Japanese boy with Waardenburg syndrome type 1 - Nature.com” plus 1 more
“A novel PAX3 mutation in a Japanese boy with Waardenburg syndrome type 1 - Nature.com” plus 1 more |
| A novel PAX3 mutation in a Japanese boy with Waardenburg syndrome type 1 - Nature.com Posted: 03 Mar 2016 12:00 AM PST AbstractWaardenburg syndrome type 1 (WS1) is a rare autosomal dominant disorder characterized by hair hypopigmentation, abnormal iris pigmentation, and congenital hearing loss. WS1 is caused by mutations in paired box gene 3 (PAX3). We identified a novel PAX3 mutation (c.1107 C>G, p.Ser369Arg) in a Japanese WS1 patient showing abnormal right iris pigmentation, right-sided congenital hearing loss, synophrys, incomplete left cleft lip, and cryptorchidism. Mutations in paired box gene 3 (PAX3, NM_181457.3.) are responsible for Waardenburg syndrome type 1 (WS1; OMIM 193500),1,2 which is clinically characterized by hair hypopigmentation, abnormal iris pigmentation, and congenital hearing loss. Its prevalence is estimated to be 1/42,000. Furthermore, it is responsible for 1–3% of total congenital deafness.3 Diagnosis of WS1 is established by clinical findings in most individuals. Five major white forelock and hair hypopigmentation are one pair, dsytopia canthorum and W index>1.95 are one pair. (• congenital hearing loss • white forelock, hair hypopigmentation • abnormal iris pigmentation • dystopia canthorum, W index>0.95 • affected first-degree relative) and minor diagnostic criteria for WS have been proposed by the Waardenburg Consortium.4 An individual must have two major criteria or one major plus two minor criteria to be diagnosed with WS.4,5PAX3 is one of the nine human PAX genes encoding DNA-binding transcription factors.6PAX3 has 10 exons and encodes a paired box by exons 2–4 and a homeobox by exons 5 and 6.6 The patient was a 10-month-old Japanese boy born at 33 weeks of gestation with a (very low) birth weight of 1,250 g. He was referred to our hospital because of an incomplete left cleft lip. Body weight at the time of the first visit was 6,700 g at 10 months of age. Although his physique was petite, his nutritional status was good. The patient showed pigmentation abnormalities of the right iris, ocular hypertelorism (W index: 2.00), right-sided congenital hearing loss, synophrys, incomplete left cleft lip, and cryptorchidism. He was diagnosed with WS1 on the basis of clinical findings and the criteria proposed by the Waardenburg Consortium.4 According to the parents, his family members, including the proband and mother, showed clinical features consistent with WS1 (Figure 1). The proband is indicated by an arrow (Figure 1a:21). His mother's hair was speckled with grey. However, she showed no other symptoms (Figure 1a:20). His mother's aunt showed a lateral iris pigmentation abnormality (Figure 1a:14), and his mother's elder sister showed a lateral iris pigmentation abnormality and bilateral congenital hearing loss (Figure 1a:19). 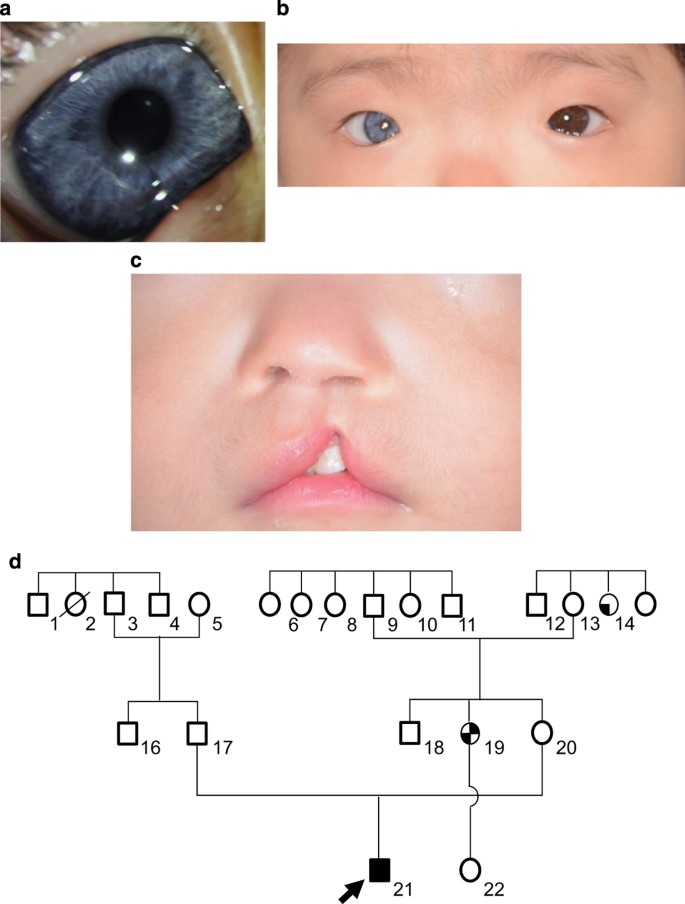 (a) Family pedigree of the WS1 patient. Squares indicate males and circles indicate females. Shading in these symbols corresponds to symptoms as follows: upper left quadrant, ocular hypertelorism; lower left, abnormal iris pigmentation; upper right, hearing loss; and lower right, synophrys. (b) Abnormal pigmentation of the right iris of the patient. (c) Ocular hypertelorism (W index: 2.00) of the patient. (d) Incomplete left cleft lip of the patient. Surgery to repair the cleft lip was performed at 14 months, at a weight of 6,900 g. The preoperative assessment was not problematic. His parents wanted the patient to undergo a genetic test for PAX3. For the genetic diagnosis, blood samples were obtained from the patient after informed consent was obtained from the parents. Genomic DNA was extracted from the blood samples using a standard protocol. All 10 exons and exon–intron junctions of the PAX3 gene were amplified from genomic DNA by PCR using primers designed with primer software (Genetyx software, Genetyx, Shibuya, Japan). The primers were designed to include an intron of 50 bp surrounding the exon. The sequencing was performed using forward and reverse primers. All the primer sequences are available on request. Sequencing was performed with a BigDye Terminator v3.1 Cycle Sequencing Kit and a 3500×l Genetic Analyzer capillary sequencer (Life Technologies, Carlsbad, CA, USA). Mutation analysis revealed a novel missense mutation (c.1107 C>G, p.Ser369Arg) in exon 7 (Figure 2a). 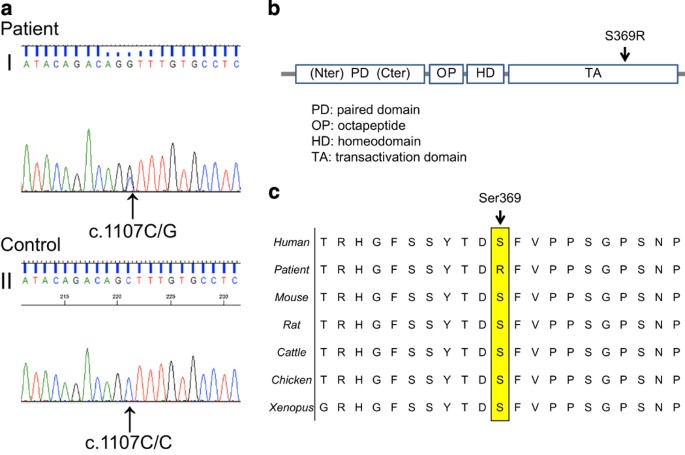 (a) Sequence chromatograms of the WS1 patient and a control. I: A heterozygous change, c.1107C>G, was identified in the affected boy. II: Healthy controls are wild-type at this position. (b) Schematic diagram of the PAX3 gene. The arrow indicates the mutated gene. The reported missense mutation identified in the WS1 patient is presented. (c) Cross-species multiple alignment of PAX3 protein sequences, showing evolutionary conservation of the altered amino acid, Ser369. The mutation was predicted to be pathogenic using PolyPhen-2 (probably damaging, Hum Var 0.958, Hum Div 0.776).7 MutationTaster indicated that the mutation was a disease-causing mutation (model: simple_aae, prob: 0.999980746176575).8 Parental analysis was refused. The mutated Ser369 residue is evolutionarily conserved from fish to humans (Figure 2b). Ser369 resides in a highly conserved transactivation domain that interacts with DNA (Figure 2c). These symptoms are believed to arise due to the mutation, which prevents the formation of a covalent disulfide bond (S–S bond), resulting in protein instability. The variant p.Ser369Arg is located in exon 7. To date, other WS-related mutations have been reported in this exon.9–12 Most of the reported mutations in PAX3 are localized in exons 2–6. Deletions comprise two whole gene deletions and two intragenic deletions of exon 7, and exons 8 and 9.12,13 There is no correlation between the mutation type (missense, nonsense, or deletion) and severity of the phenotype.13 A few mutations of PAX3 have been analyzed for their functional consequences.1,2,13 In addition, functional studies are required to assess the exact molecular mechanisms induced by the c.1107C>G mutation. Cleft lip and palate are low-penetrance features of WS1 and have been found in 2.8% of the reported cases.14 In conclusion, we identified a novel PAX3 mutation (c.1107 C>G, p.Ser369Arg) in a Japanese boy showing pigmentation abnormality in the right iris, right-sided congenital hearing loss, synophrys, incomplete left cleft lip, and cryptorchidism due to WS1. This finding further broadens the existing knowledge of mutations of the PAX3 gene in WS1 patients, enabling clinicians and genetic counsellors to offer better clinical services. WS1 pathogenesis should be taken into account when treating WS1 infants with a cleft lip. ReferencesReferences
Data Citations
Author informationEthics declarationsCompeting interestsThe authors declare no conflict of interest. Rights and permissionsThis work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ About this articleCite this articleYoshida, Y., Doi, R., Adachi, K. et al. A novel PAX3 mutation in a Japanese boy with Waardenburg syndrome type 1. Hum Genome Var 3, 16005 (2016) doi:10.1038/hgv.2016.5 Further reading
|
| Many rare diseases are linked to hearing loss - Healthy Hearing Posted: 15 May 2019 12:00 AM PDT Edith Veitch, now 17, was 10 when an audiologist diagnosed her hearing loss and fitted her with aids.  rare disease that causes hearing loss. Her mother, Margo, a community college instructor in Illinois, is unsure when the hearing loss set in. "We don't have [hearing loss] in the family so we weren't looking for it," she said. In school, because Edith has an intellectual disability, she had been assigned a companion to repeat things to her in class. Her family then opted for a deaf and hard-of-hearing program—the classes were small and it wasn't clear how quickly her hearing might deteriorate. Many of her classmates have cochlear implants or rely mainly on ASL. But, Edith's hands are small and it is difficult for her fingers to form shapes. "When her peers sign to her, she was signing back, but she has become more self-conscious," Veitch said. Eventually her family learned that Edith has Myhre syndrome, a rare disorder of the connective tissue. Fortunately, as with many rare diseases, hearing aids have been an effective treatment. "She appreciates being able to hear," Veitch said of her daughter's hearing aids. Many rare diseases can cause hearing lossScientists have identified 7,000 diseases, like Myhre syndrome, that are considered rare. As defined in the U.S by the Orphan Drug Act of 1983, rare diseases each affect fewer than 200,000 people. However, up to 30 million Americans live with a rare disease. Many, but not all, have been traced at least in part to genes, with signs that appear at birth or early in life. At least 400 rare syndromes include hearing loss as a symptom, according to BabyHearing.org. They can lead to different types of hearing loss, the main types being sensorineural and conductive.
The degree of loss can vary widely from person to person. For some people, hearing aids will be sufficient. For others, cochlear implants and/or learning American Sign Language will be recommended. In many cases, a rare disease can cause multiple anatomical and functional changes in the ears. A prime example of this is Turner syndrome. Hearing loss may be apparent at birth or soon afterBecause of state programs aided by the federal government, nearly all American babies have a hearing test within the first month of life. About two or three out of every 1,000 newborns in the U.S. have a detectable hearing loss in one or both ears. More: What parents need to know about newborn hearing screening The hearing loss may be a sign of a rare disease. Babies with Mondini dysplasia, for example, are born with one and a half coils in the cochlea instead of the standard two, in either one or both ears. Most children with this condition have profound hearing loss. They may need a surgical repair, as well as a cochlear implant, but some can benefit from hearing aids. Babies with KID syndrome, Donnai-Barrow syndrome, and Wildervanck syndrome—among other rare diseases—may have hearing loss. Sometimes the loss is not present at birth but develops soon after. Babies with the most common and severe form of Krabbe disease develop symptoms in the first six months, which include fevers, muscle weakness and hearing and vision loss. Or it develops laterAnd sometimes, as with Edith, the hearing loss comes much later. People with Alport syndrome, for example, often lose hearing in late childhood or early adolescence and may be treated with hearing aids. Similarly, people with Alström syndrome tend to have progressive hearing loss in both ears that may begin in childhood and be treated with hearing aids. Other notable rare disorders linked to hearing loss
Symptoms can be unpredictableSymptoms can vary widely in severity among people with the same disorder. And intellectual ability may or may not be affected. For example, children with Carpenter syndrome may be of normal intelligence but it is common for them to have an intellectual disability and sometimes hearing loss. Such is the case with Myhre syndrome, the condition that Edith has. At least 80 percent of people with it have a hearing impairment, as well as intellectual disability and stiff joints. Although Edith's hearing did decline, she does not have a profound loss. She happily wears her hearing aids, even at home. She also takes an art class with 32 students, and as her mother noted, "many don't know she has hearing aids." For more informationFor more information on rare diseases, see Global Genes, the National Organization for Rare Disorders (NORD), and the Genetic and Rare Disease Information Center (GARD). If you suspect you or a loved one has hearing loss, visiting our large online directory of consumer-reviewed hearing clinics is a good next step. |

| You are subscribed to email updates from "waardenburg syndrome" - Google News. To stop receiving these emails, you may unsubscribe now. | Email delivery powered by Google |
| Google, 1600 Amphitheatre Parkway, Mountain View, CA 94043, United States | |



Comments
Post a Comment